Epidermolysis bullosa types
 Fact-checked by
Fact-checked by Epidermolysis bullosa (EB) is a group of rare skin conditions that cause the skin to be very fragile and blister easily. In most cases, they are genetic skin disorders that are present from birth, with symptoms ranging from mild to life-threatening, depending on the specific type and subtype.
Knowing which type of EB a person has is important for providing effective medical care, anticipating possible complications, and planning for long-term care.
Major types of EB
EB comprises a group of several skin blistering disorders that are typically classified based on the location and genetic cause of the blisters forming within skin layers. All types cause the skin to become fragile, but differ in their severity, symptoms, and long-term outcomes.
There are many different forms of EB, but the four main types are:
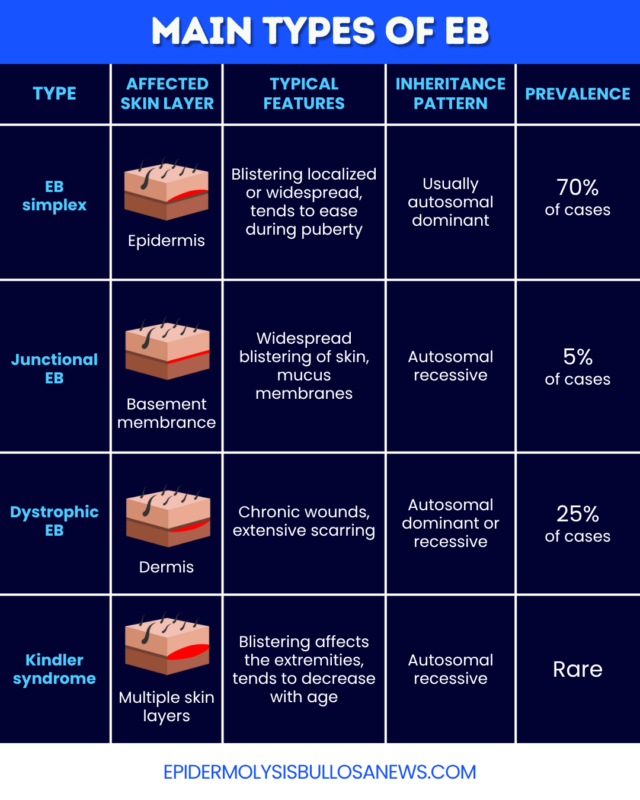
- EB simplex (EBS): The most common and usually the mildest type. Blisters form in the upper layer of the skin.
- Junctional EB (JEB): A rarer and more severe type. Blistering may affect the skin and mucus membranes lining internal parts of the body.
- Dystrophic EB (DEB): The second most common form of EB. Causes deep blistering and scarring, which can be moderate to severe.
- Kindler syndrome: A rare form of EB. Blistering occurs in multiple skin layers.
Within most of these major types, there are additional EB subtypes that differ in severity and clinical features, depending on the specific genetic mutations and other factors.
Besides these genetic forms of EB, there is also an autoimmune disease called EB acquisita (EBA), in which the immune system mistakenly attacks a skin structural protein, causing symptoms similar to those of DEB.
Epidermolysis bullosa simplex (EBS)
EBS is the milder and most common type of EB, accounting for approximately 70% of cases. Usually, blistering occurs in the upper layer of the skin (epidermis), worsening with heat, sweating, or physical activity. Blisters generally heal without scarring, and may be limited to specific areas of the body, such as the hands or feet, or more widespread. There are multiple EBS subtypes that differ in the extent and severity of blistering.
EBS is caused by mutations that affect keratins, which are structural proteins found in the epidermis. These genetic mutations are usually passed down in an autosomal dominant manner, meaning that one defective gene copy from either biological parent is sufficient for a child to develop the disease.
EBS symptoms usually appear at birth or early infancy and may become more apparent as a child grows older and becomes more physically active. Blistering in EBS also tends to ease during puberty. Most people with EBS have a normal life expectancy.
Junctional epidermolysis bullosa (JEB)
JEB is a more severe and rarer EB type, accounting for approximately 5% of cases. It affects the basement membrane between the epidermis and the skin layer underneath, called dermis. It is marked by widespread blistering of the skin and mucus membranes, including those lining the inside of the mouth and eyes.
There are two main JEB subtypes: a severe subtype that causes extensive blistering, can lead to several complications, and is associated with a reduced lifespan; and an intermediate subtype that causes less severe blistering and is generally associated with a normal life expectancy.
JEB can be caused by genetic mutations affecting different proteins, including laminin-332, which are important for keeping the upper and lower layers of the skin together. These mutations are typically inherited in an autosomal recessive manner, meaning that a mutated gene copy from each biological parent is required for a child to develop JEB.
JEB symptoms typically manifest at birth, and can range from mild to life-threatening.
Dystrophic epidermolysis bullosa (DEB)
DEB is the second most common form of EB, accounting for around 25% of all cases. It is marked by blistering in the dermis and mucus membranes, which can lead to chronic wounds and extensive scarring. There are several DEB subtypes, with different inheritance patterns and degrees of severity.
DEB is caused by mutations that affect type VII collagen, a protein that plays a key role in anchoring skin layers together and maintaining skin integrity. These mutations can be inherited in an autosomal dominant or recessive manner. Recessive forms of the disease are usually more severe than dominant forms.
Due to possible complications, people with DEB require long-term monitoring. Repeated scarring may cause fusion of fingers or toes, difficulty swallowing, or nutritional deficiencies. Additionally, people with recessive DEB, especially those with more severe forms, have a higher risk of developing an aggressive type of skin cancer, which may also shorten life expectancy.
Kindler syndrome
Kindler syndrome is a rare form of EB. Blistering can occur in multiple skin layers and tends to affect the extremities. People with Kindler syndrome often have increased sensitivity to sunlight. Skin thinning or discoloration and thickening of the skin on the palms of the hands or soles of the feet are also common. Blistering tends to decrease with age.
This condition is inherited in an autosomal recessive manner and is caused by mutations in the gene encoding a protein called kindlin-1, which is important for maintaining skin integrity.
People with Kindler syndrome also have a higher risk of developing an aggressive type of skin cancer, which may reduce life expectancy.
How EB types are diagnosed
Correctly diagnosing a person’s EB type is crucial for medical providers to tailor treatment, take precautions against possible future complications, and make long-term care plans. The EB diagnosis process usually involves a combination of:
- Clinical evaluation: Doctors review symptoms, family history, and rule out other possible conditions.
- Genetic testing: Used to identify known EB-causing mutations and is considered the gold standard diagnostic method for EB.
- Skin biopsy and microscopy: Involves removing a small skin sample that is then examined under a microscope to pinpoint which skin layers and proteins are affected.
Living with different EB types
Because there is no effective cure for EB, treatment generally aims to ease EB symptoms, prevent complications, and improve patients’ quality of life. The type and level of care depend on the specific EB type, subtype, and symptom severity.
For milder forms of EB, this may involve taking measures to protect the skin and prevent blistering and infections. Severe forms of EB require a higher level of care, including consistent wound cleaning and dressing, nutritional support, and pain management.
Common challenges associated with daily life with EB include pain, infection, limited mobility, and emotional stress for both patients and caregivers. Educational and financial resources offered by patient or advocacy groups are available to support patients and caregivers, making living with EB easier.
Epidermolysis Bullosa News is strictly a news and information website about the disease. It does not provide medical advice, diagnosis, or treatment. This content is not intended to be a substitute for professional medical advice, diagnosis, or treatment. Always seek the advice of your physician or other qualified health provider with any questions you may have regarding a medical condition. Never disregard professional medical advice or delay in seeking it because of something you have read on this website.